Introduction of ChIP-Seq
Chromatin immunoprecipitation sequencing (ChIP-Seq) stands as a cornerstone in contemporary genomics, revered for its prowess in mapping intricate protein-DNA interactions across the genome. Integrating the precision of chromatin immunoprecipitation (ChIP) with the robust throughput of next-generation sequencing (NGS), this method enables precise localization of DNA binding sites for transcription factors and other DNA-associated proteins [1].
Initiating with in vivo cross-linking of proteins to DNA, ChIP-Seq captures dynamic protein-DNA interactions within cellular chromatin. Subsequent fragmentation of chromatin, achieved typically through sonication or enzymatic digestion, yields manageable DNA segments. Immunoprecipitation with antibodies specific to the protein of interest enriches these protein-bound DNA fragments, which are subsequently purified and primed for sequencing.
The application of next-generation sequencing technologies then facilitates the sequencing of millions of short DNA fragments. Alignment of these sequences to a reference genome unveils comprehensive maps of protein binding sites across the genome, enabling detailed exploration of transcription factor binding motifs, histone modifications, and other regulatory elements pivotal in gene regulation mechanisms.
The transformative impact of ChIP-Seq extends across diverse realms of biological inquiry. In epigenetics, it has been instrumental in charting genome-wide distributions of histone modifications, offering crucial insights into their regulatory roles in gene expression. In cancer biology, ChIP-Seq has pinpointed aberrant binding sites of oncogenic transcription factors, shedding light on mechanisms underlying tumorigenesis. Additionally, in developmental biology, ChIP-Seq has unraveled intricate transcriptional networks orchestrating cellular differentiation and developmental pathways.
This methodological fusion of specificity and high-throughput sequencing has redefined our ability to decode genomic complexity, offering researchers unprecedented avenues to elucidate fundamental biological processes and disease mechanisms.
The Principle and Process of ChIP-Seq
The principle of ChIP-Seq: Under physiological conditions, H3the DNA and protein in the cells are crosslinked (Crosslink) and then the cells are lysed, the chromosomes are separated, the chromatin is randomly cut by ultrasound or enzyme treatment, and the DNA fragments bound to the target protein are precipitated by the specific recognition reaction of antigen and antibody. The DNA fragments bound to the protein are then released through reverse crosslink (di-crosslink), and finally the sequence of the DNA fragments is obtained by sequencing [2, 3].
The step 1: Cross-Linking and Fragmentation
The initial stage of ChIP-Seq commences with the treatment of cells using formaldehyde, facilitating the cross-linking of proteins to DNA. This chemical process preserves the intricate interactions between proteins and DNA within the chromatin structure. Subsequently, the cross-linked chromatin undergoes fragmentation into smaller fragments, typically ranging from 200 to 600 base pairs, achieved through ultrasonic waves. This fragmentation step ensures that the DNA segments bound by the protein of interest are in appropriately sized fragments for subsequent analytical procedures.
This process underscores the foundational step of ChIP-Seq, integrating chemical cross-linking with physical fragmentation to prepare chromatin for further analysis. This methodological approach is pivotal in elucidating the precise genomic locations where proteins interact with DNA, thus advancing our understanding of gene regulation mechanisms.
The step 2: Immunoprecipitation
Subsequently, an antibody specific to the target protein is employed to selectively enrich the DNA-protein complexes. The high specificity of the antibody guarantees the isolation of DNA fragments exclusively bound to the protein of interest. These complexes are then precipitated from the solution, facilitating the separation of the target-bound DNA from the remaining chromatin constituents.
This step in the ChIP-Seq protocol exemplifies the precision required to discern and capture specific DNA-protein interactions. By selectively enriching the targeted complexes, researchers can effectively isolate and analyze DNA sequences associated with particular proteins, thereby advancing our insights into gene regulatory mechanisms.
The step 3: Purification and Library Preparation
Subsequently, the protein-DNA complexes undergo reverse cross-linking to disentangle DNA from proteins. The resulting purified DNA fragments undergo PCR amplification to bolster their quantity. Following amplification, these fragments are meticulously prepared for high-throughput sequencing through the construction of a sequencing library. This crucial step entails the addition of adapters to the ends of the DNA fragments, essential for facilitating the sequencing process.
This methodical approach in ChIP-Seq underscores the meticulous preparation required to transform protein-DNA interactions into sequenced data. By amplifying and preparing these fragments with precision, researchers ensure accurate and comprehensive analysis of genomic binding sites, offering invaluable insights into intricate biological processes.
The step 4: High-Throughput Sequencing
The prepared DNA library undergoes sequencing using next-generation sequencing (NGS) technologies, yielding millions of short sequencing reads. These reads collectively depict the DNA fragments that were specifically bound by the protein of interest.
This sequencing step in ChIP-Seq is pivotal for capturing and decoding the genomic locations where proteins interact with DNA. By generating a vast array of sequence data, NGS enables comprehensive analysis of protein-DNA interactions across the genome, thereby advancing our understanding of gene regulation mechanisms and biological processes.
The step 5: Data Analysis
In the concluding step, researchers meticulously align these sequence reads to a reference genome. This methodical mapping process enables precise identification of the DNA locations where the target protein binds across the entire genome. By correlating these sequence reads with established genomic sequences, researchers attain comprehensive insights into the specific sites of interaction between proteins and DNA. This genome-wide mapping plays a pivotal role in elucidating complex gene regulatory mechanisms and enhancing our understanding of molecular-level biological processes.
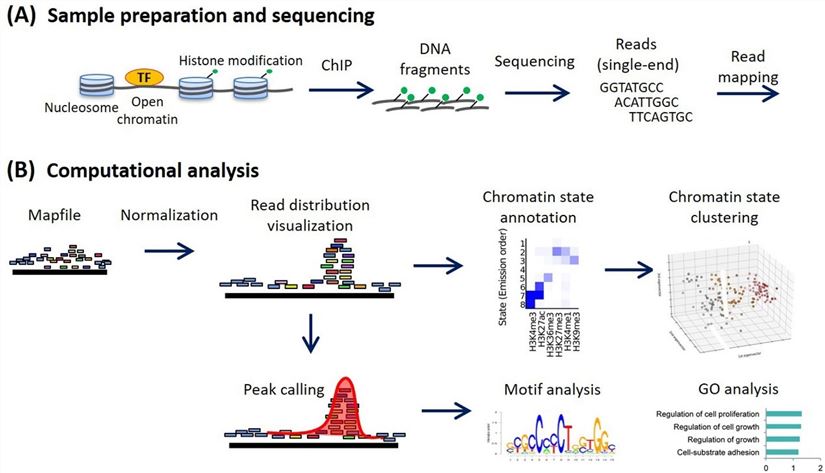
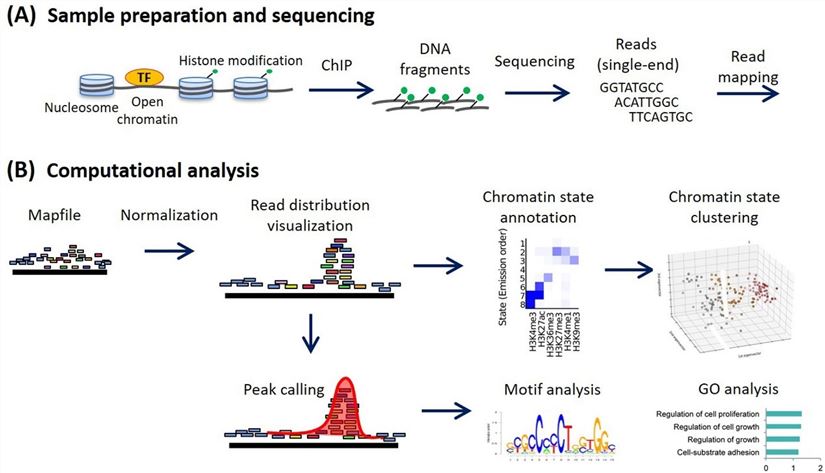
Figure 1. ChIP-seq analysis workflow (Ryuichiro Nakato, Toyonori Sakata, 2021). (A) Sample preparation and sequencing. (B) Computational analysis in a canonical ChIP-seq analysis. Various analyses are implemented using normalized read distribution.
Advantages of ChIP-Seq: Unraveling the Genomic Mysteries
Enhanced Resolution and Coverage
ChIP-Seq stands out for its exceptional resolution and coverage, surpassing its predecessor, ChIP-chip. Research demonstrates that ChIP-Seq achieves base-pair resolution, enabling precise mapping of DNA-binding sites. This heightened resolution is crucial for identifying subtle yet biologically significant peaks that may be obscured in array-based methods. Furthermore, ChIP-Seq’s capability to survey the entire genome without constraints imposed by fixed probe sequences enhances exploration of repetitive regions and heterochromatin.
Noise Reduction and Increased Sensitivity
ChIP-Seq minimizes inherent noise associated with hybridization-based techniques like ChIP-chip. By reducing complexities such as cross-hybridization in nucleic acid interactions, ChIP-Seq yields cleaner and more precise data. This enhanced sensitivity enables detection of nuanced protein-DNA interactions that might be overshadowed in array-based assays.
Dynamic Range and Linearity
ChIP-Seq exhibits a compelling dynamic range and maintains linear signal responses, distinguishing it from array-based methods prone to non-linearities. This characteristic is pivotal for accurately quantifying protein-DNA binding affinities and deciphering intricate regulatory mechanisms.
Computational Analysis and Data Interpretation
The wealth of data generated by ChIP-Seq necessitates robust computational tools for analysis. Advances in bioinformatics algorithms have significantly bolstered our capacity to extract meaningful insights and unravel complex biological pathways from ChIP-Seq data. State-of-the-art techniques such as sophisticated peak calling and differential peak analysis, coupled with integration across various omics datasets, enhance ChIP-Seq’s utility in elucidating gene regulatory networks.
learn more: DNA Methylation Sequencing Methods